

Proteins analysis using polyacrylamide gel electrophoresis (PAGE)
Electrophoresis is the process of migration of charged molecules to the oppositely charged electrodes under the influence of an electric field. The velocity with which the charged particles migrate in a constant electric field is termed electrophoretic mobility (μ). This rate of migration depends on the intrinsic properties like net charge (q), shape and size of the biomolecule, and external parameters like the voltage gradient of the electric field (E), and resistance offered by the medium in the form of friction (f).
$$ \mu = \frac{v}{E} =\frac{q}{f}$$
Proteins are made of amino acids that have –COO and –NH2 groups and side chains that assume charge at different pH. Other than their isoelectric point (or pH at which the net charge is 0), proteins are charged molecules and undergo migration towards the oppositely charged electrodes in an electric field. This is the principle of gel electrophoresis to separate protein molecules with differential charge/mass.
Electrophoresis using gels made from a polymer of acrylamide, cross-linked by bisacrylamide (in the ratio 29:1) is referred to as Polyacrylamide gel electrophoresis (PAGE). These gels are chemically inert and stable. The co-polymerization of acrylamide monomers with cross-linking reagent N-N’-methylene bisacrylamide gives a clear transparent gel with pores. This polymerization process is a free radical catalysis initiated by Ammonium persulfate (APS) and catalyzed by N, N, N’N’-Tetramethylenediamine (TEMED) (Figure 1). TEMED speeds up the decomposition of the persulfate ion to give a free radical.
S2O8 2- + e- → SO4 2- + SO4 -
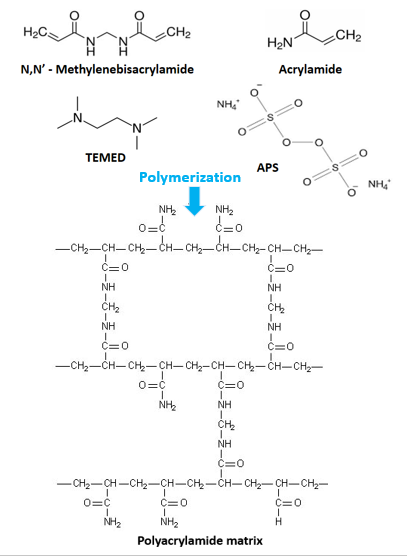
Figure 1: The polymerization of acrylamide and bisacrylamide to form the polyacrylamide gel used in PAGE.
The overall mobility of the protein samples in the polyacrylamide gel is proportional to the pore size, which is a function of the concentration of acrylamide (%T) as well as that of bisacrylamide (%C). The pore size decreases with an increase in the acrylamide content (%T). Based on the molecular weight of the proteins to be analysed, gels of varying acrylamide content (hence different pore size) are used.
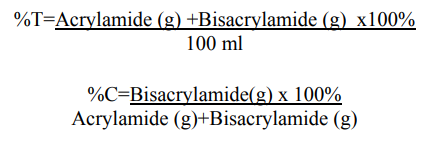
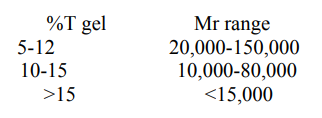
PAGE can be performed in reducing (with the anionic detergent SDS) or non-reducing conditions. In native or non-denaturing gel electrophoresis SDS is not used and the proteins retain their native structure and enzymatic activity. Here, the separation of proteins is influenced by its the net charge, shape, and size. SDSPAGE, on the other hand, is the most widely used gel electrophoretic technique where SDS strongly binds to the protein samples and impart a uniform negative charge so that the separation is purely based on the differences in mass. SDS binds to the hydrophobic regions disrupts all the interactions in the folded protein, unfolds and linearizes it by binding at intervals of every 2 residues. The reducing agents Beta-mercaptoethanol or dithiotritol (DTT) helps to break disulphide (-S-S-) linkages in the protein. So, in the gel monomers are seen as distinct bands corresponding to their molecular weights (Figure 2).
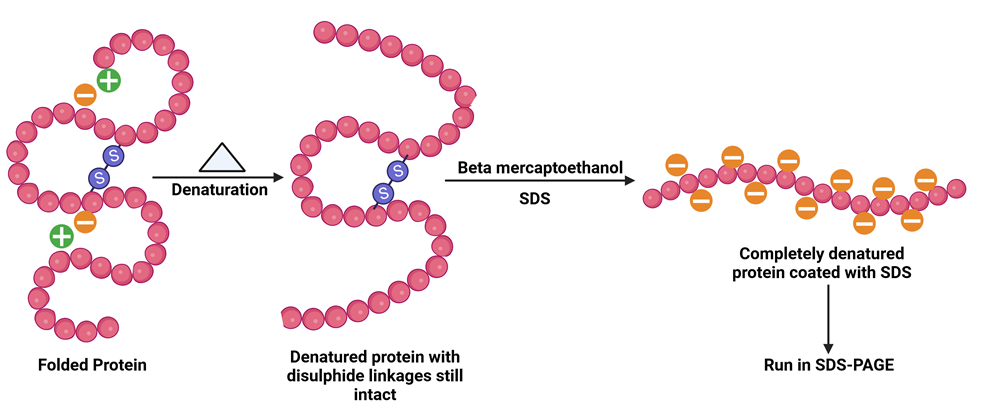
Figure 2: The principle of SDS-PAGE
The molecular weights of the protein samples can be determined from their relative mobility (as shown in Figure 3) using SDS-PAGE.
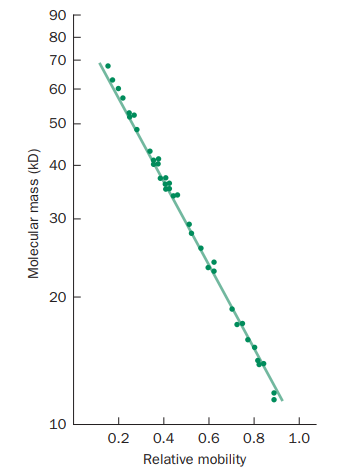
Figure 3: Determination of molecular weight of proteins using SDS-PAGE
To improve the resolution, a discontinuous system of stacking and resolving gel is used. The stacking gel concentrates all the protein samples into a single band before entering into the resolving gel where the separation begins. The differences between the ionic strength and pH of the buffers render different properties to the stacking and resolving gels. The former has high porosity and a Tris-Cl buffer pH of 6.8, the latter has smaller pores and a Tris-Cl buffer pH of 8.8 (Figure 4).
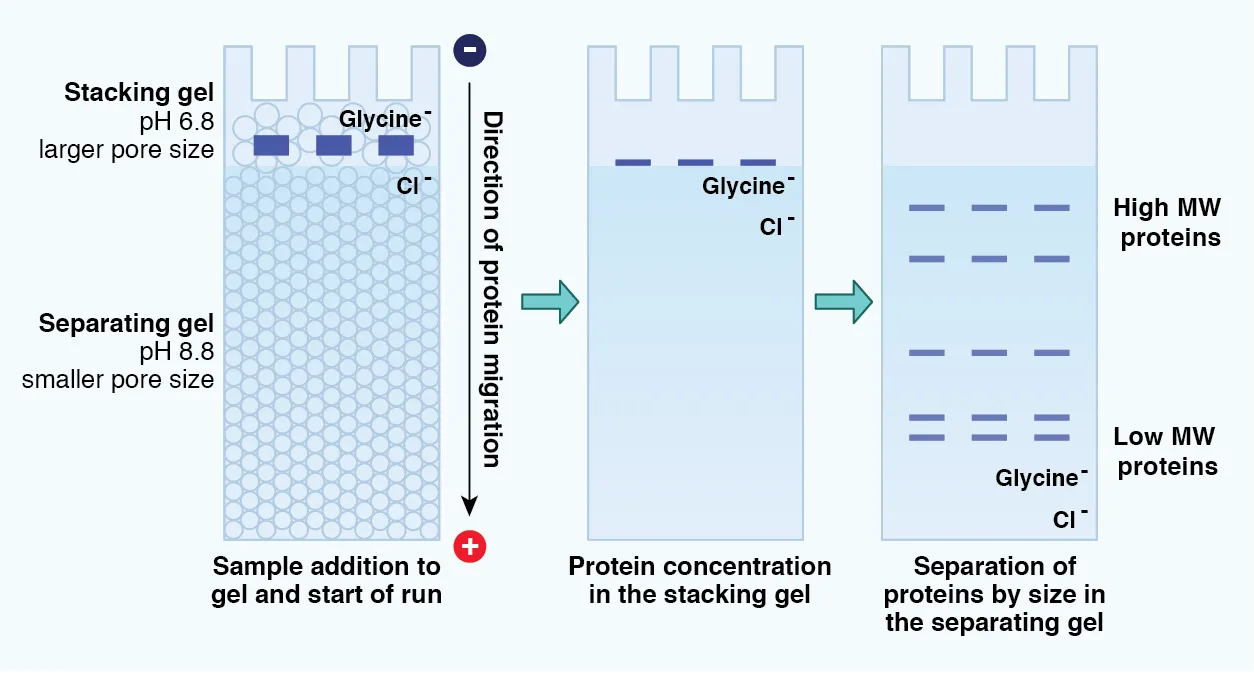
Figure 4: Determination of molecular weight of proteins using SDS-PAGE.
At the stacking gel, the counter ion (glycine) remains neutral (zwitter ion), and chloride ions act as the principal current carriers and migrate towards the anode. The negatively charged SDS-protein complexes are stacked as a thin band between glycine and chloride ions. In the resolving gel, glycine forms glycinate ions and follows the chloride ions, the SDS-protein molecules are well resolved by the molecular sieving effect of the pores based on their size difference.
For visualization, Bromophenol blue and Xylene Cyanol are used to track the progress of the electrophoretic process. Coomassie brilliant blue staining and Silver staining (for 100 times better sensitivity) are used for final visualization. Coomassie stain preferably binds to the basic residues in a protein like Histidine, Lysine and Arginine to form a protein-dye complex. In silver staining, silver ions are reduced to metallic silver at sites occupied by the proteins based on differences in the redox potential.